
Mitochondria in Health and Disease
 Mitochondria are the cell’s source of energy and play an essential role in the proper functioning of cells and organs. Healthy mitochondria generate adenosine triphosphate (ATP), the energy molecule required by our cells and organs to function properly. Mitochondria also produce a minimal level of reactive oxygen species (ROS) as secondary products of respiration.
Mitochondria are the cell’s source of energy and play an essential role in the proper functioning of cells and organs. Healthy mitochondria generate adenosine triphosphate (ATP), the energy molecule required by our cells and organs to function properly. Mitochondria also produce a minimal level of reactive oxygen species (ROS) as secondary products of respiration.
When mitochondria dysfunction, ATP production is reduced and ROS production is increased, with an accompanying increase in oxidative stress. The increased ROS that dysfunctional mitochondria produce can damage multiple cellular components (including DNA, proteins, lipids and the mitochondria themselves), which results in the disruption of normal biological functioning and disease.
Mitochondrial dysfunction is believed to arise either as a result of inherited genetic defects or through damage-signals encountered during disease. In either case, the result is the generation of excessive ROS and reduced ATP production. Dysfunctional mitochondria are a key element in a variety of serious diseases (both rare and common), and thus a promising therapeutic target.
Mitochondrial Medicines
The development of Mitochondrial Medicines is challenged by the difficulty of targeting the mitochondria (passing both the cellular membrane and the mitochondrial outer membrane) without affecting the mitochondrial membrane potential or causing mitochondrial toxicity.
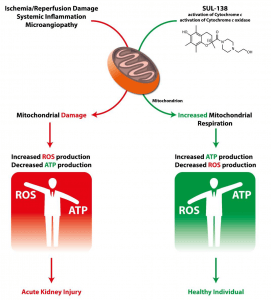
SUL-138 appears to target and protect dysfunctional mitochondria without affecting healthy mitochondria and cells. SUL-138, in preclinical models, is a mitochondrial protective agent that improves cell viability and organ function in several disease models including renal, metabolic, pulmonological and hypothermia-associated disorders.
SUL-138 Mechanism of Action
SUL-138 is formulated for systemic intravenous infusion, but oral delivery formulations are currently under development with promising results.
In preclinical models, SUL-138 is uniquely able to reach the mitochondria, where it restores electron transport to improve cellular energy production and reduce oxidative stress created by the over-production of ROS.
SUL-138 functions via direct reduction of Cytochrome c and the maintenance of the enzyme Cytochrome c oxidase in the mitochondrial inner membrane. Extensive preclinical investigations have revealed that SUL-138, through its novel mechanism:
a. Maintains mitochondrial bioenergetics under pathological conditions (including ultrastructure, membrane potential and respiration);
b. Increases ATP levels under pathological conditions; and
c. Decreases ROS production under pathological conditions.
These novel mechanisms ameliorates electron transport deficiencies in disease. Given these innovational mechanisms-of-action, SUL-138 can provide therapeutic benefits in mitochondrial disease, such as Acute Kidney Injury.
Mitochondria in Surgery-related Acute Kidney Injury
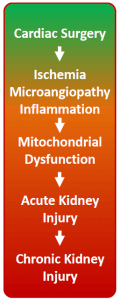 Acute kidney injury (AKI) is a critical clinical condition with no effective treatment. AKI incidence continues to increase and AKI is associated with high mortality at short and long-term, increased risk of progression to chronic kidney disease and end-stage renal disease. Cardiac bypass surgery is a common cause of AKI in humans with incidences as high as 30%. Survivors of AKI are plagued by a variety of chronic sequelae contributing to decreased quality of life and increased utilization of healthcare resources.
Acute kidney injury (AKI) is a critical clinical condition with no effective treatment. AKI incidence continues to increase and AKI is associated with high mortality at short and long-term, increased risk of progression to chronic kidney disease and end-stage renal disease. Cardiac bypass surgery is a common cause of AKI in humans with incidences as high as 30%. Survivors of AKI are plagued by a variety of chronic sequelae contributing to decreased quality of life and increased utilization of healthcare resources.
The initiation of AKI following cardiac bypass surgery is multifactorial and poorly understood, however ischemia-reperfusion (I/R) injury is pivotal. Hampered kidney perfusion – due to reduced cardiac output, increased systemic inflammation, hemodynamic alterations, volume depletion and perioperative administration of nephrotoxic drugs – contributes to I/R-induced AKI following cardiac surgery. Acute kidney injury (AKI) is defined by an acute rise in serum creatinine levels or decrease in urine output. As the bulk of renal cell mass is composed of tubular cells. Tubular cell death or injury is the main morphological feature of AKI. Tubular cells are extremely rich in mitochondria and mitochondrial abnormalities are a key feature of AKI.
This mitochondrial injury progresses into mitochondrial dysfunction, resulting in the inability to generate ATP and excessive ROS production (oxidative stress) and, thus, leading to cell injury. As a consequence mitochondria are therapeutic targets in the treatment of renal disease.
Mitochondrial dysfunction is a key pathophysiological element in AKI during both the initiation and recovery phases. Ischemia results in rapid tubular mitochondrial fragmentation and shortening. The mitochondrial membrane potential and mitochondrial respiration decrease while mitochondrial ROS increases. This results in ATP depletion, lipid peroxidation, and the release of proapoptotic proteins. Reperfusion, or kidney reoxygenation, results in rapid restoration of mitochondrial respiration and a burst in ROS production. However, the mitochondria remain impaired in their ATP synthesizing function. Thus, ATP recovery is often delayed, which inhibits the initiation of energy-dependent cellular repair mechanisms, and contributes to persistence of AKI.
SUL-138 maintains mitochondrial bioenergetics under pathological conditions such as ischemia/reperfusion and thus counteracts the processes that underlie the formation of AKI, i.e. tubular mitochondrial dysfunction. Maintenance of ATP generation under ischemic conditions and decreased ROS formation upon reperfusion protect the tubular cells from injury, whereas accelerated ATP formation would allow for repair of damage encountered during the ischemia-reperfusion phase.